The Future of Drug Discovery

Two New Technologies Overcome Hurdles in Drug Development
Drug development and obstacles are nearly synonymous, since one must carefully balance target effectiveness with normal tissue toxicity. Drug development is facing several major hurdles, but thanks to two new innovative technologies, drug development is ready to take another leap forward.
Robert Hromas, M.D., FACP, dean of the Joe R. and Teresa Lozano Long School of Medicine at UT Health San Antonio, believes that the two technologies, Proteolysis Targeting Chimeras (PROTACs) and active site covalent inhibitors will go a long away in clearing these obstacles.
Dr. Hromas, a hematologic oncologist, said his interest in drug development for cancer started some 15 years ago. “About half of blood cancers are caused by chromosomal translocations, and I studied the effects of those chromosomal translocations in leukemia for a decade,” he says. “About 15 years ago, I asked myself a question that changed my career: What causes these chromosomal translocations in the first place?”
His search for an answer became personal as his father dealt with chronic myeloid leukemia, a disease caused by a single chromosomal translocation. If chromosome 9 and 22 both break, chromosome 9 erringly ligates itself with 22, forming a fusion gene BCR-ABL, and this mutant protein tells the bone marrow stem cells to divide without stopping, causing this leukemia.

Many mutated proteins that cause cancer are “undruggable,” Dr. Hromas explains, because it is impossible to find a chemical that can bind to and inhibit them. Either the protein is too small, and there is no chemical “pocket” for a drug to fit or because inhibiting the target protein would result in too much toxicity to normal tissue.
Another obstacle occurs in proteins that have the appropriate structure to be druggable. Target cells are adept at overcoming inhibitors over time. For example, cancer cells often mutate the target protein to make it resistant to the drug. “For the proteins we can drug, we’ve beat them to death,” says Dr. Hromas. “We’ve optimized chemicals that can bind to and inhibit the target to the point where we really can’t optimize the drug any further. There just isn’t much more chemical space to explore. And this is true for drug development in many diseases, not just cancer.”
PROTACs
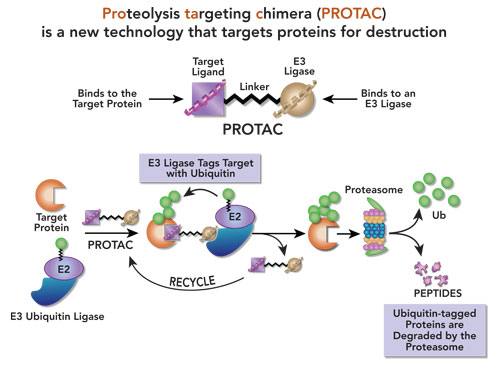
PROTAC drug technology overcomes both of these barriers in a novel approach. A PROTAC consists of a chemical that will bind and interact with a protein and then target it for degradation by the cell’s own digestive machinery. The ligand that binds to the target protein is linked to a second chemical that will bind to a conjugating enzyme family called E3 ligases. These two linked ligands make up the “chimera.” The PROTAC pulls the E3 ligase enzyme next to the target protein and then attaches a ubiquitin molecule to it, which assigns it for digestion by the cell, in the proteasome. Unlike an inhibitor, Dr. Hromas explains, “a PROTAC will target a protein for degradation. So, you are pulling the E3 ligase against your target and it attaches a ubiquitin molecule to the target. That ubiquitin molecule is like a red flag on a tree in the forest and then the logging company comes by and cuts it down.”
The proteasome chews up the targeted protein into amino acids to be rebuilt in other proteins. “You can get ten-fold higher effectiveness using a PROTAC because it is catalytic and not inhibitory, and so it can be recycled within the cells,” says Dr. Hromas. “Further, it doesn’t rely on occupancy of an active site of an enzyme that causes cancer for its activity. Rather, it is continuously degrading that target protein that causes cancer, and it stays inside the cell functioning long after it has gone from the bloodstream.”
Another advantage of PROTACs is that an E3 ligase can be selected that is specific to the cancer cell, thereby reducing toxicity to normal cells. Dr. Hromas’ and colleagues Drs. Doahong Zhou and Guangrong Zheng at the University of Florida demonstrated this in a Nature Medicine paper. They selected an E3 ligase that targets the cancer survival protein BCL-XL present in many T-cell malignancies and other cancers. “It’s one of the most famous undruggable targets in cancer research because inhibiting it kills normal platelets, causing bleeding.”
 “BCL-XL tells cancer cells to survive forced rapid division,” Dr. Hromas said. In healthy cells, any forced rapid division is subject to natural apoptosis (cellular suicide). “But that doesn’t happen in many cancers because they over express this immortality protein BCL-XL,” he says. By choosing an E3 ligase, in this case the Von Hippel-Lindau “VHL” E3 ligase which is poorly expressed in platelets, but highly active in the cancer cells, BCL-XL can be effectively targeted for destruction while platelets are left unharmed, because platelets have low levels of VHL. Once this survival protein is degraded, the cancer cells become much more sensitive to chemotherapy drugs.
“BCL-XL tells cancer cells to survive forced rapid division,” Dr. Hromas said. In healthy cells, any forced rapid division is subject to natural apoptosis (cellular suicide). “But that doesn’t happen in many cancers because they over express this immortality protein BCL-XL,” he says. By choosing an E3 ligase, in this case the Von Hippel-Lindau “VHL” E3 ligase which is poorly expressed in platelets, but highly active in the cancer cells, BCL-XL can be effectively targeted for destruction while platelets are left unharmed, because platelets have low levels of VHL. Once this survival protein is degraded, the cancer cells become much more sensitive to chemotherapy drugs.
PROTAC technology is not limited to cancer, Dr. Hromas says, but could also target congestive heart failure, Alzheimer’s disease, or virtually any disease in which you can target a protein that causes the disease. “We’re targeting T-cell lymphoma for our first clinical trial simply because about half of those require BCL-XL for survival and current drugs are less effective, working in only about one-third of cases.”
COVALENT INHIBITORS
Another exciting area in drug development is covalent inhibitors. Inhibitors aim to prevent an enzyme from functioning by binding to the reactive site of an enzyme. “The inhibitors we’re using today are about as potent as you can get in many diseases,” Dr. Hromas says. “We’re at the end of chemical space here. That means all the drug modifications possible have already been made. To overcome that, we can now create a reactive group on the inhibitory drug that covalently links to the inside of that active site on the targeted enzyme such that it cannot be washed out.” In a covalent bond, atoms in the enzyme and the chemical inhibitor share electrons, and so they cannot be separated. This makes the inhibitor far more potent and decreases the chance that resistance will develop.
The only way a cancer cell can overcome these new approaches, Dr. Hromas says, “is to synthesize new proteins as replacements, which is a slow process and tough for the cancer cell to do, and if it does, we can re-dose.” Thus, using either a PROTAC or a covalent inhibitor can overcome the barriers to drug development previously described. A PROTAC can be used to target an “undruggable” cancer protein because it does not have to bind to the protein’s active site. Binding anywhere on the target protein can be sufficient to target it for ubiquitination and degradation. In addition, by carefully choosing the E3 ligase that performs the ubiquitination for low expression in normal tissue but high in cancer, one can reduce normal tissue toxicity. In addition, turning a regular inhibitor into a covalent inhibitor can increase drug effectiveness and decrease development of resistance in cancer cells.
As always, new obstacles remain in generating PROTACs and covalent inhibitors to potentially target other cancerous and non-cancerous diseases like Alzheimer’s and congestive heart failure, but Dr. Hromas is optimistic. “I think drug development is going to move heavily in the direction of these two principles.” The team approach to drug development at the medical school will lead the way. “In the last 18 months, we’ve recruited a dozen well-funded, high-impact scientists who are on the cutting edge of their fields. Just coming to work every day and being around brilliant people who are really hard-working and care deeply about their patients and their students is extremely rewarding.” It is this team that has identified multiple novel cancer targets. The team uses the Center for Innovation in Drug Discovery (CIDD), a joint venture between UT Health San Antonio and UTSA, to discover ligands that bind to these target proteins from which to synthesize PROTACs and covalent inhibitors.
Progress has been remarkable, with a number of compounds heading for clinical trials in the next few years. Thus, for even the most discouraged cancer patients, whose cancers have become resistant to all therapies, new hope is developing at UT Health.




